

Introduction
Sepsis refers to a systemic inflammatory response syndrome caused by infection, which can lead to multiple organ dysfunction [1, 2]. Acute kidney injury (AKI) is an independent risk factor for increased death risk in sepsis patients [3, 4]. Sepsis can promote the release of inflammatory cytokines including interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6), leading to apoptosis and an inflammatory response of kidney cells, resulting in AKI [5, 6]. Therefore, it is of great significance to strengthen the treatment of sepsis patients with AKI to improve their prognosis.
Long non-coding RNA (lncRNA) generally refers to RNA molecules that do not have protein coding capabilities and the transcript length exceeds 200 nucleotides [7, 8]. A large number of studies have suggested that lncRNA is abnormally expressed in various diseases and is involved in the regulation of disease progression, including sepsis-induced AKI [9, 10]. For example, lncRNA TUG1 was downregulated in sepsis-induced AKI patients, and overexpressed TUG1 could inhibit the AKI cell model injury induced by lipopolysaccharide (LPS) in vitro [11]. Moreover, lncRNA PVT1 silencing could alleviate LPS- induced AKI cell apoptosis and the inflammatory response via regulating the nuclear factor-κB (NF-κB) signaling pathway [12]. Therefore, lncRNA might be a key target for the treatment of sepsis-induced AKI.
LncRNA nuclear enriched abundant transcript 1 (NEAT1) is a widely expressed lncRNA in many diseases [13]. Previous studies had indicated that NEAT1 was highly expressed in ischemia/reperfusion-induced AKI patients, and that the absence of NEAT1 could relieve CoCl2-induced AKI cell injury by suppressing cell apoptosis [14]. In an LPS-induced AKI cell model, NEAT1 could regulate the activity of the NF-κB signaling pathway to promote cell injury through targeting miR-204 [15]. However, the mechanism of NEAT1 in regulating the progression of AKI has not been fully elucidated and is worth further exploration.
Functionally, lncRNA has been proven to be a competitive endogenous RNA (ceRNA) of microRNA (miRNA) to relieve the inhibition of miRNA on downstream genes [16]. MiR-22-3p is a miRNA with significantly low expression in sepsis patients and has been shown to regulate the progression of sepsis-induced AKI [17, 18]. CXC chemokine ligand 12 (CXCL12), also known as stromal cell-derived factor 1 (SDF-1), belongs to the subfamily of CXC chemokines. The results of Zhang et al. showed that miR-20a suppressed apoptosis and the release of inflammatory cytokines (IL-1β, TNF-α and IL-6) in LPS-induced HK2 cells by inhibiting the NF-κB signaling pathway via targeting CXCL12 [19].
Interestingly, we found that the expression of NEAT1 and miR-22-3p had a significant negative correlation in the serum of patients with sepsis. According to the prediction of bioinformatics analysis software, miR-22-3p had binding sites with NEAT1 and CXCL12, so we assumed the existence of the NEAT1/miR-22-3p/CXCL12 axis. In this study, we constructed an LPS-induced kidney cell injury model and cecal ligation and puncture (CLP) mouse model to clarify the role of NEAT1 in sepsis-induced AKI, and confirmed the presence of the NEAT1/miR-22-3p/CXCL12 axis through a series of experiments.
Material and methods
Serum samples
The blood samples were collected from 32 sepsis patients and 32 healthy volunteers recruited from emergency department and EICU ward of Ruijin Hospital. All patients with sepsis were confirmed by the hospital to be in critical care according to the Sequence Organ Failure Assessment (SOFA) score. The SOFA score of all sepsis patients is greater than 2. After centrifugation, the serum was collected and stored at –20°C. For this study, all patients and volunteers signed written informed consent. This study was approved by the Ethics Committee of Ruijin Hospital.
Cell culture and LPS treatment
The human kidney tubular epithelial cell line (HK2) and the embryonic kidney cell line (HEK293) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM medium (Gibco, Grand Island, NY, USA) supplemented with 1% Penicillin-Streptomycin (Biological Industries, Kibbutz Beit Haemek, Israel) and 10% fetal bovine serum (FBS; Gibco) at 37°C in an incubator with 5% CO2. LPS (Solarbio, Beijing, China) was used to induce cell injury in vitro. Briefly, HK2 cells were treated with different concentrations (0, 2.5, 5, and 10 µg/ml) of LPS for 12 h to screen the optimal treatment concentration, and treated with 10 µg/ml LPS for different times (0, 4, 8 and 12 h) to screen the optimal treatment time.
Cell transfection
When cells reached 50% confluences, cell transfection was performed. NEAT1 small interference RNA (si-NEAT1) and its negative control (si-NC), pcDNA NEAT1 or CXCL12 overexpressing vector (NEAT1 or CXCL12) and its negative control (pcDNA) were synthesized by Ribobio (Guangzhou, China). The miR-22-3p mimic and inhibitor (miR-22-3p and anti-miR-22-3p) as well as their negative controls (miR-NC and anti-miR-NC) were obtained from General Biosystems (Anhui, China). Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) was used to transfect them into cells. After transfection for 12 h, transfected HK2 cells were treated with 10 µg/ml LPS for 12 h.
Quantitative real-time PCR
The RNA was isolated using TRI Reagent (Sigma- Aldrich, St. Louis, MO, USA), and cDNA was generated using a cDNA Reverse Transcription Kit (Multisciences, Hangzhou, China). A SYBR Green PCR Kit (Takara, Dalian, China) was used to perform quantitative real-time PCR (qRT-PCR). Relative expression was normalized with GAPDH or U6 and analyzed by the 2-ΔΔCT method. Primer sequences are shown in Table 1.
Table 1
Primer sequences used in this study
Cell counting kit 8 assay
After treatment or transfection, HK2 cells were harvested and reseeded into 96-well plates. After incubation for 48 h, 10 µl of cell counting kit 8 (CCK8) solution (Medchemexpress, Monmouth Junction, NJ, USA) was pipetted into each well and the plates were incubated for 4 h. Then, the viability of cells was measured at 450 nm using a microplate reader.
Flow cytometry
After treatment or transfection, HK2 cells were digested with trypsin and collected into a centrifuge tube. After washing with PBS 3 times, the cells were resuspended with 1 × binding buffer. Then, the cell suspension was stained with Annexin V-FITC and propidium iodide using the Annexin V-FITC Apoptosis Detection Kit (eBioscience, San Diego, CA, USA). The apoptosis rate was detected using a flow cytometer equipped with CellQuest software.
Enzyme-linked immunosorbent assay
Interleukin 1β, TNF-α and IL-6 enzyme-linked immunosorbent assay (ELISA) kits were bought from Invitrogen. According to the manufacturer’s instructions, the concentrations of IL-1β, TNF-α and IL-6 were examined by corresponding ELISA kits, respectively.
Dual-luciferase reporter assay
StarBase v2.0 tool (http://starbase.sysu.edu.cn/starbase2/index.php) was used to predict the binding sites between miR-22-3p and NEAT1 or CXCL12 3'UTR. The fragments of wild-type (WT) and mutant-type (MUT) NEAT1 were amplified and cloned into the pmirGLO vector (Promega, Madison, WI, USA) to build the WT-NEAT1 and MUT-NEAT1 vectors, respectively. Similarly, the CXCL12 3'UTR-WT and CXCL12 3'UTR-MUT vectors were generated in the same way. The vectors were co-transfected with the miR-22-3p mimic or miR-NC into HEK293 cells using Lipofectamine 3000. Afterwards, the Dual-Luciferase Reporter Assay System (Promega) was used to detect the luciferase activity of each vector after transfection for 48 h.
RNA immunoprecipitation assay
This assay was performed using an RNA immunoprecipitation (RIP) Kit (BersinBio, Guangzhou, China). In brief, HK2 cells were collected and lysed using polysome lysis buffer. After removal of DNA using DNase salt stock and DNase, the cell lysates were incubated with Ago2 or IgG (Abcam, Cambridge, MA, USA) at 4°C for 16 h, followed by incubating with protein A/G magnetic beads at 4°C for 1 h. The RNA was extracted from the beads complex and the expression levels of NEAT1, miR-22-3p and CXCL12 were detected by qRT-PCR.
Western blot analysis
Total protein was extracted by RIPA lysate buffer (Solarbio). The same amount of protein was separated by 10% SDS-PAGE gel and transferred to PVDF membrane (Roche, Basel, Switzerland). The membrane was blocked using 5% skim milk, and then incubated with the primary antibodies (Arigobio, Hsinchu City, Taiwan) including anti-CXCL12 (1 : 10,000), P65 (1 : 3,000), phosphorylated-P65 (p-P65; 1 : 1,000), IκB-α (1 : 2,000) or phosphorylated-IκB-α (p-IκB-α; 1 : 1,000) or anti-GAPDH (1 : 8,000) at 4°C overnight. Then, the membrane was incubated with Goat anti-Rabbit or Mouse secondary antibody (1 : 20,000; Arigobio) for 2 h, and then the protein signals were detected using West Femto ECL Substrate (Solarbio).
Animal experiments
Male C57BL/6 mice were purchased from Beijing HFK Bioscience Co., Ltd. (Beijing, China) and randomly divided into 4 groups (n = 5/group). AKI mouse models were constructed by performing CLP surgery as previously described [20]. The packaged lentivirus short hairpin NEAT1 (sh-NEAT1) and its control (sh-NC) were used for injection via the tail vein. After 24 h, mice were sacrificed, and the blood and kidney tissues were collected for detecting. The animal study was approved by the Animal Ethics Committee of Shanghai Jiao Tong University School of Medicine.
Statistical analysis
GraphPad Prism 5.0 software (GraphPad, La Jolla, CA, USA) was used for statistical analyses. The data are shown as mean ± standard deviation. Comparisons were measured using Student’s t-test or one-way analyses of variance. The correlation between miR-22-3p and NEAT1 or CXCL12 expression was evaluated using Pearson correlation analysis. P < 0.05 indicated statistical significance.
Results
NEAT1 was highly expressed and miR-22-3p was lowly expressed in sepsis patients and LPS-induced HK2 cells
In the serum of sepsis patients, we discovered that NEAT1 was markedly upregulated compared with that in the serum of healthy volunteers (Fig. 1A). In contrast, the expression of miR-22-3p was lower in the serum of sepsis patients than in healthy volunteers (Fig. 1B). We found that the expression levels of NEAT1 and miR-22-3p in the serum of sepsis patients showed a significant negative correlation (Fig. 1C).
Fig. 1
Expression of NEAT1 and miR-22-3p in sepsis patients. Relative NEAT1 (A) and miR-22-3p (B) expression levels in the serum of sepsis patients and healthy volunteers were examined by qRT-PCR. C) Pearson correlation analysis was used to analyze the correlation between NEAT1 and miR-22-3p. *p < 0.05

In the LPS-treated HK2 cell model, we found that the expression of NEAT1 was significantly increased in a dose-dependent and time-dependent manner (Fig. 2A, B), while the expression of miR-22-3p was remarkably decreased in a dose-dependent manner and time-dependent manner (Fig. 2C, D).
Fig. 2
Expression of NEAT1 and miR-22-3p in LPS-induced HK2 cells. After being treated with different concentrations (0, 2.5, 5, and 10 μg/ml) of LPS for 12 h, the expression levels of NEAT1 (A) and miR-22-3p (C) in HK2 cells were detected by qRT-PCR. After being treated with 10 μg/ml LPS for different times (0, 4, 8, and 12 h), the expression levels of NEAT1 (B) and miR-22-3p (D) in HK2 cells were examined by qRT-PCR. *p < 0.05

Knockdown of NEAT1 promoted viability, and suppressed apoptosis and the inflammatory response in LPS-induced HK2 and HEK293 cells
To explore the function of NEAT1, we silenced NEAT1 expression in HK2 and HEK293 cells. The decreased NEAT1 expression revealed that the transfection efficiency of si-NEAT1 was good (Fig. 3A and Supplementary Fig. 1A). Further experiments showed that LPS could suppress the viability and enhance the apoptosis rate of HK2 and HEK293 cells, while NEAT1 knockdown could reverse this effect (Fig. 3B, C and Supplementary Fig. 1B, C). Furthermore, LPS markedly increased the concentrations of IL-1β, TNF-α and IL-6, indicating that LPS accelerated the inflammatory response of HK2 and HEK293 cells. However, silenced NEAT1 significantly repressed the concentrations of IL-1β, TNF-α and IL-6 in LPS-induced HK2 and HEK293 cells (Fig. 3D and Supplementary Fig. 1D).
Fig. 3
Effect of NEAT1 silencing on LPS-induced HK2 cell injury. HK2 cells were transfected with si-NC or si-NEAT1, and then treated with 10 μg/ml LPS for 12 h. A) Relative NEAT1 expression was detected by qRT-PCR to assess the transfection efficiency. Cell viability and apoptosis rate were determined using CCK8 assay (B) and flow cytometry (C). D) ELISA assay was used to test the concentrations of IL-1β, TNF-α and IL-6. *p < 0.05

NEAT1 directly interacted with miR-22-3p
To reveal the mechanism by which NEAT1 regulated LPS-induced HK2 cell injury, bioinformatics analysis was used to predict the miRNAs that could interact with NEAT1. The results from the StarBase v2.0 tool showed that miR-22-3p had complementary binding sites with NEAT1 (Fig. 4A). To confirm the interaction between them, dual-luciferase reporter assay and RIP assay were carried out. The results showed that the miR-22-3p mimic could reduce the luciferase activity of the WT-NEAT1 reporter vector, but had no effect on the luciferase activity of the MUT-NEAT1 reporter vector (Fig. 4B). Moreover, miR-22-3p and NEAT1 could be enriched in Ago2 compared with IgG (Fig. 4C). For further exploring the regulation of NEAT1 on miR-22-3p, we constructed the pcDNA NEAT1 overexpression vector. The increased NEAT1 expression confirmed that the transfection of the pcDNA NEAT1 overexpression vector was successful (Fig. 4D). Then, we detected miR-22-3p expression and found that NEAT1 silencing remarkably enhanced miR-22-3p expression in LPS-induced HK2 cells, while NEAT1 overexpression had an opposite effect (Fig. 4E).
Fig. 4
NEAT1 sponged miR-22-3p. A) The sequences of WT-NEAT1 and MUT-NEAT1 are shown. The interaction between miR-22-3p and NEAT1 was confirmed using dual-luciferase reporter assay (B) and RIP assay (C). D) Expression of NEAT1 was measured by qRT-PCR to verify the transfection efficiency of pcDNA NEAT1 overexpression vector. E) Relative miR-22-3p expression was detected by qRT-PCR in LPS-induced HK2 cells transfected with si-NC, si-NEAT1, pcDNA or NEAT1. *p < 0.05
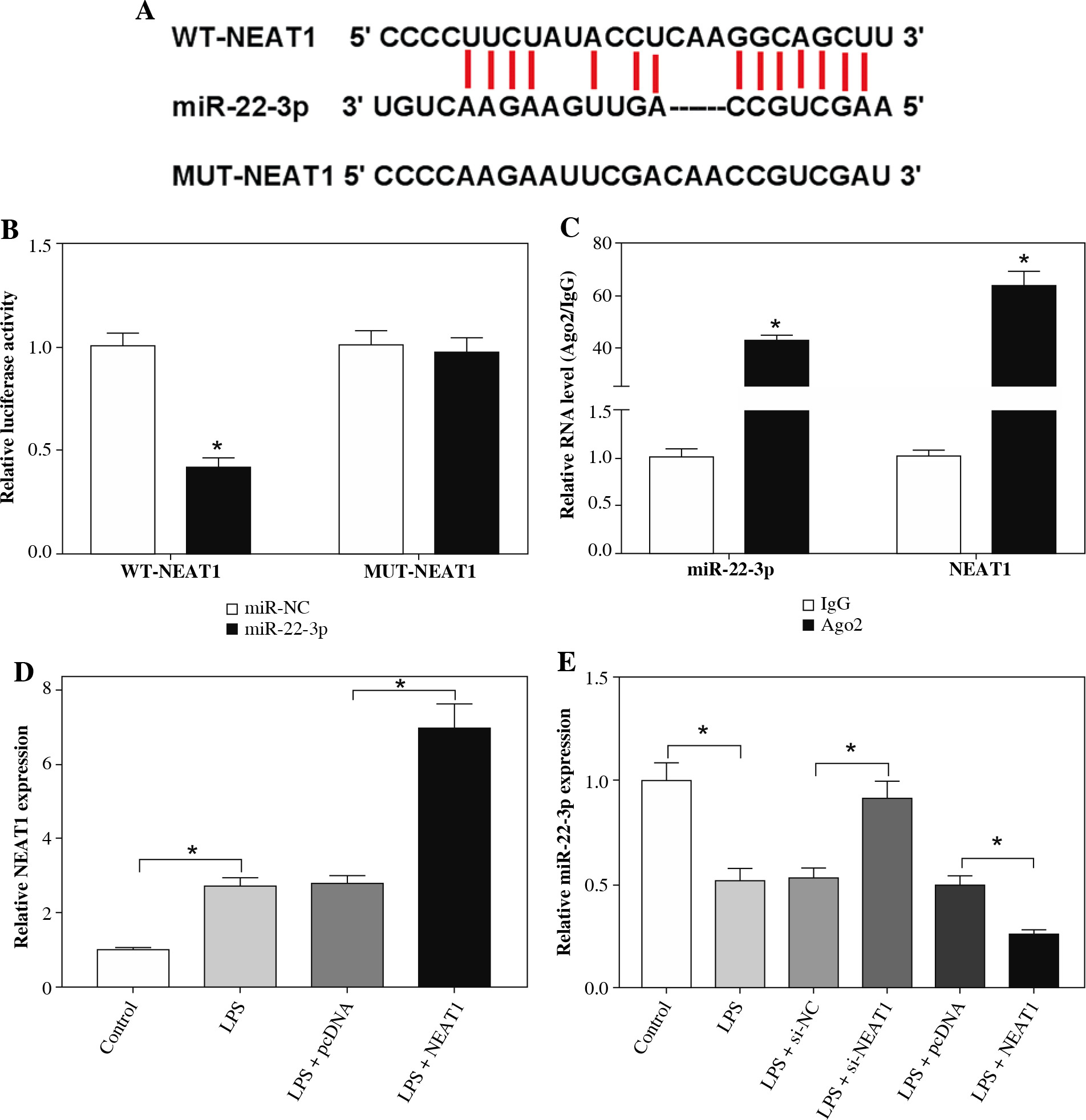
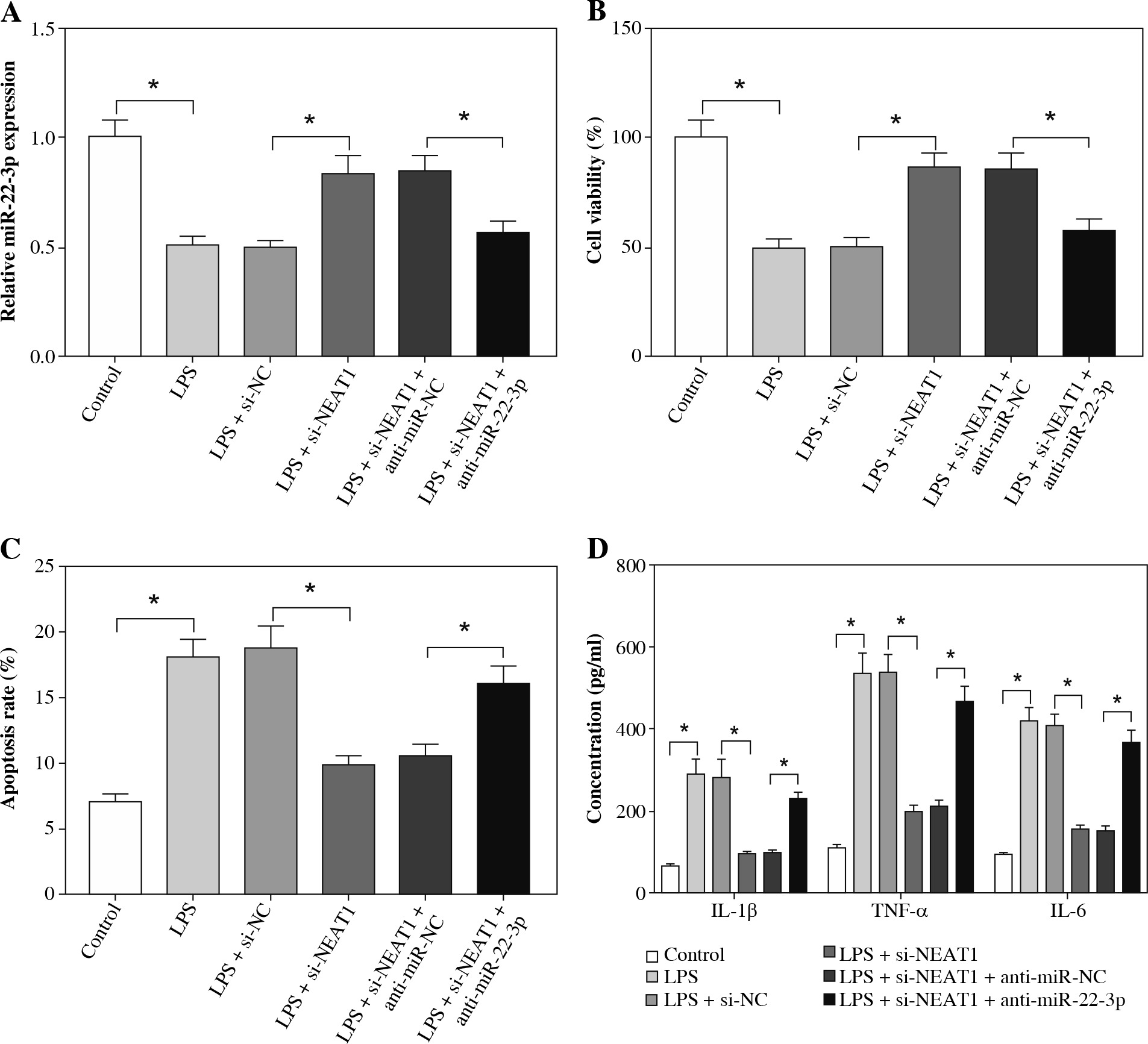
MiR-22-3p inhibiter reversed the inhibition effect of NEAT1 silencing on LPS-induced HK2 cell injury
Further experiments were used to explore whether NEAT1 regulated LPS-induced kidney cell injury by regulating miR-22-3p. Through measuring the expression of miR-22-3p in LPS-induced HK2 cells, we confirmed that the transfection of si-NEAT1 and anti-miR-22-3p was successful (Fig. 5A). As shown in Figure 5B-D, we discovered that miR-22-3p inhibitor could reverse the promotion effect of NEAT1 silencing on viability and the suppressive effect of it on apoptosis rate and the concentrations of inflammatory cytokines in LPS-induced HK2 cells. All data suggested that NEAT1 negatively regulated miR-22-3p expression to mediate LPS-induced HK2 cell injury.
Fig. 5
Effects of NEAT1 silencing and miR-22-3p inhibiter on LPS-induced HK2 cell injury. HK2 cells were transfected with si-NC, si-NEAT1, si-NEAT1 + anti-miR-NC or si-NEAT1 + anti-miR-22-3p, and then treated with 10 μg/ml LPS for 12 h. A) Relative miR-22-3p expression was measured by qRT-PCR to evaluate the transfection efficiency. CCK8 assay (B) and flow cytometry (C) were performed to test cell viability and apoptosis rate. D) Concentrations of IL-1β, TNF-α and IL-6 were determined using ELISA assay. *p < 0.05
MiR-22-3p targeted CXCL12
To study the mechanism of miR-22-3p, we selected the candidate targets of miR-22-3p using the StarBase v2.0 tool. As shown in Figure 6A, CXCL12 3'UTR was found to have targeted binding sites with miR-22-3p. The luciferase activity of the CXCL12 3’UTR-WT vector rather than the CXCL12 3 ’UTR-MUT vector could be repressed by the miR-22-3p mimic (Fig. 6B). In addition, miR-22-3p and CXCL12 could be markedly enriched in Ago2 (Fig. 6C). Furthermore, compared to healthy volunteers, the expression of CXCL12 in the serum of sepsis patients was significantly increased (Fig. 6D). In LPS-treated HK2 cells, we also discovered that the CXCL12 protein expression was remarkably promoted in a dose-dependent manner and time-dependent manner (Fig. 6E, F). Additionally, CXCL12 expression was negatively correlated with miR-22-3p expression in sepsis patients (Fig. 6G). Therefore, we concluded that CXCL12 was a target of miR-22-3p.
Fig. 6
CXCL12 was a target of miR-22-3p. A) The fragments of CXCL12 3’UTR-WT and CXCL12 3’UTR-MUT are exhibited. Dual-luciferase reporter assay (B) and RIP assay (C) were used to confirm the interaction between miR-22-3p and CXCL12. D) Relative CXCL12 mRNA expression in the serum of sepsis patients and healthy volunteers was assessed by qRT-PCR. E) After treatment with different concentrations (0, 2.5, 5, and 10 μg/ml) of LPS for 12 h, the CXCL12 protein expression in HK2 cells was examined by Western blot (WB) analysis. F) After treatment with 10 μg/ml LPS for different times (0, 4, 8, and 12 h), the CXCL12 protein expression in HK2 cells was detected by WB analysis. G) The correlation between CXCL12 and miR-22-3p was evaluated using Pearson correlation analysis. *p < 0.05

CXCL12 reversed the suppressive effect of miR-22-3p on LPS-induced HK2 cell injury
In addition, we explored whether CXCL12 participated in the regulatory effect of miR-22-3p on LPS-induced kidney cell injury. By detecting the protein expression of CXCL12, we found that the miR-22-3p mimic could inhibit the expression of CXCL12, while CXCL12 overexpression could reverse the inhibition effect of the miR-22-3p mimic on CXCL12 expression (Fig. 7A). MiR-22-3p overexpression could enhance the viability and restrain the apoptosis of LPS-induced HK2 cells, while overexpressed CXCL12 could reverse this effect (Fig. 7B, C). In addition, the inhibition effect of miR-22-3p on the concentrations of IL-1β, TNF-α and IL-6 could also be reversed by CXCL12 overexpression (Fig. 7D). Hence, these data suggested that miR-22-3p regulated LPS-induced HK2 cell injury by regulating CXCL12 expression.
Fig. 7
Effects of miR-22-3p and CXCL12 overexpression on LPS-induced HK2 cell injury. HK2 cells were transfected with miR-NC, miR-22-3p, miR-22-3p + pcDNA or miR-22-3p + CXCL12, and then treated with 10 μg/ml LPS for 12 h. A) Relative CXCL12 protein expression was tested using WB analysis to assess the transfection efficiency. CCK8 assay (B) and flow cytometry (C) were used to determine cell viability and apoptosis rate. D) ELISA assay was performed to measure the concentrations of IL-1β, TNF-α and IL-6. *p < 0.05

The NEAT1/miR-22-3p/CXCL12 axis regulated LPS-induced HK2 cell injury by regulating the activity of the NF-κB signaling pathway
To confirm the regulatory effect of NEAT1 on CXCL12 expression, the CXCL12 expression was tested in LPS-induced HK2 cells co-transfected with si-NEAT1 and anti-miR-22-3p. The results shown in Figure 8A revealed that NEAT1 knockdown markedly suppressed CXCL12 expression and the miR-22-3p inhibitor could reverse this effect, indicating that CXCL12 expression was negatively regulated by NEAT1. Additionally, NF-κB is a classical signaling pathway involved in cellular responses to stimuli [21]. In order to explore whether the activity of the NF-κB signaling pathway was affected by the NEAT1/miR-22-3p/CXCL12 axis in LPS-induced HK2 cells, the relative expression of p-P65 and p-IκB-α was detected. As shown in Figure 8B, we confirmed that LPS could remarkably increase the protein expression of p-P65 and p-IκB-α in HK2 cells. Furthermore, NEAT1 silencing significantly inhibited the protein expression of p-P65 and p-IκB-α in LPS-induced HK2 cells, while this effect could be reversed by the miR-22-3p inhibitor or CXCL12 overexpression (Fig. 8C). All the results indicated that the activity of the NF-κB signaling pathway was regulated by the NEAT1/miR-22-3p/CXCL12 axis in LPS-induced HK2 cells.
Fig. 8
The NEAT1/miR-22-3p/CXCL12 axis regulated activity of the NF-κB signaling pathway. A) Protein expression of CXCL12 was measured by Western blot (WB) analysis in LPS-induced HK2 cells transfected with si-NC, si-NEAT1, si-NEAT1 + anti-miR-NC or si-NEAT1 + anti-miR-22-3p. B) After treatment with 10 μg/ml LPS for 12 h, the protein expression levels of p-P65 and p-IκB-α in HK2 cells were detected by WB analysis. C) The protein expression levels of p-P65 and p-IκB-α were examined using WB analysis in LPS-induced HK2 cells transfected with si-NC, si-NEAT1, si-NEAT1 + anti-miR-NC, si-NEAT1 + anti-miR-22-3p, si-NEAT1 + pcDNA or si-NEAT1 + CXCL12. *p < 0.05

NEAT1 knockdown improved the inflammatory response in CLP-induced AKI mice
To further confirm our conclusion, we performed the animal experiments. The results showed that NEAT1 was overexpressed in CLP-induced AKI mouse models, while it was decreased in CLP models injected with sh-NEAT1 (Supplementary Fig. 2A). In CLP models, the serum levels of creatinine, BUN and NGAL were markedly reduced after knockdown of NEAT1 (Supplementary Fig. 2B-D). Moreover, downregulated NEAT1 also suppressed the concentrations of IL-1β, TNF-α, and IL-6 in CLP models (Supplementary Fig. 2E). In addition, we observed that miR-22-3p expression was enhanced and CXCL12 protein expression was decreased in CLP models after NEAT1 knockdown (Supplementary Fig. 2F, G). Therefore, we confirmed that NEAT1 knockdown alleviated the inflammatory response in CLP-induced AKI mice through the miR-22-3p/CXCL12 pathway.
Discussion
Acute kidney injury is a common clinical critical disease caused by sharply declining kidney function [3, 4]. The pathogenesis of sepsis-related AKI is very complex and has not been fully elucidated so far. However, released inflammatory cytokines and increased apoptosis in kidney cells are the direct causes of AKI [22, 23]. In our research, we studied the role of NEAT1 in the pathophysiology of AKI caused by sepsis. We found that NEAT1 had increased expression in sepsis patients. In LPS-induced AKI cell models and CLP-induced AKI mouse models, NEAT1 expression also was markedly promoted. Furthermore, loss-of-function assay results indicated that NEAT1 silencing could enhance viability, and inhibit apoptosis and the inflammatory response in LPS-induced HK2 and HEK293 cells, suggesting that NEAT1 knockdown could relieve LPS-induced kidney cell injury. Importantly, interference of NEAT1 also relieved the inflammatory response of CLP mice in vivo. These data illuminated that targeted inhibition of NEAT1 might be a potential treatment method for sepsis-induced AKI.
The mechanism by which lncRNA can act as a sponge for miRNA has been confirmed by many studies [24, 25]. Consistent with the previous study [17, 18], miR-22-3p expression was found to be downregulated in sepsis patients in this study. In LPS-induced AKI cell models and CLP- induced AKI mouse models, we discovered that miR-22-3p expression was negatively regulated by NEAT1. Bioinformatics analysis and further experimental verification revealed that NEAT1 could be used as a sponge for miR-22-3p, and NEAT1 regulated LPS-induced kidney cell injury via targeting miR-22-3p. Hu et al. found that miR-22-3p could inhibit the inflammatory injury of retinal pigment epithelium [26]. Recently a study revealed that miR-22-3p inhibited the inflammatory response of an LPS-induced sepsis cell model and CLP mouse model through reducing PTEN [27]. Similarly, our study indicated that miR-22-3p could accelerate viability, and hinder apoptosis and the inflammatory response in LPS-induced kidney cells. These observations revealed that miR-22-3p had anti-apoptotic and anti-inflammatory effects on LPS-induced kidney cells, and confirmed that miR-22-3p might be an important target for sepsis-induced AKI.
CXCL12 is commonly bound to specific receptors and has an essential role in angiogenesis, lymphatic system development, and the inflammatory response [28, 29]. Here, we confirmed that CXCL12 was a target for miR-22-3p. Consistent with the previous study [19], we confirmed the high CXCL12 expression in LPS-induced AKI cell models and its overexpression promoted cell apoptosis and inflammation. The rescue experiments also revealed that miR-22-3p played anti-apoptotic and anti-inflammatory roles in kidney injury by targeting CXCL12. More importantly, the positive regulatory effect of NEAT1 on CXCL12 expression was also confirmed in an LPS-induced sepsis cell model and a CLP mouse model. In addition, by detecting the expression of p-P65 and p-IκB-α, we found that NEAT1, miR-22-3p and CXCL12 could regulate the activity of the NF-κB signaling pathway, which indicated that the NEAT1/miR-22-3p/CXCL12 axis regulated LPS-induced kidney cell injury mainly by regulating the activity of the NF-κB signaling pathway.
Conclusions
In short, our results revealed that NEAT1 knockdown alleviates LPS-induced kidney cell injury through regulating the miR-22-3p/CXCL12/NF-κB pathway, which indicated that NEAT1 might be a potential target for treating sepsis-induced AKI. Our findings revealed the key functional mechanism of NEAT1 in AKI cell models and might provide new strategies for AKI treatment.